
When a generic drug hits the shelf, you might assume it’s just a cheaper copy of the brand-name version. But behind that simple label is a rigorous scientific process designed to prove it works exactly the same way in your body. That process is called a bioequivalence study. These aren’t just lab tests-they’re carefully controlled human trials that measure how your body absorbs and processes a drug. If the results don’t match the original drug within strict limits, the generic won’t get approved. Here’s how it actually works, step by step.
Why Bioequivalence Studies Exist
Before 1984, companies making generic drugs had to run full clinical trials to prove they worked. That meant years of testing, millions of dollars, and fewer generic options. The U.S. Hatch-Waxman Act changed that. It created a shortcut: if a generic drug behaves the same in the body as the brand-name drug, you don’t need to re-prove it works for every condition. You just need to prove it delivers the same amount of medicine at the same speed. That’s bioequivalence. The same logic applies in Europe, Japan, Canada, and most other countries. The goal? Safe, affordable access to medicines. The FDA estimates generics saved the U.S. healthcare system over $1.6 trillion between 2010 and 2019.Step 1: Choose the Right Reference Drug
Every study starts with the brand-name drug-the reference listed drug (RLD). You can’t compare a generic to just any version of the brand. You need the exact one approved by regulators. Typically, companies select a single batch from one of three production lots, ideally one with a mid-range dissolution profile. This ensures you’re testing against a representative version of the original. In Japan, regulators require the reference product to be the same one sold on the market. In the U.S., the FDA mandates the RLD be the one listed in the Orange Book. Getting this wrong means your entire study is invalid.Step 2: Prepare the Test Product
The generic drug you’re testing must be made at commercial scale-not a lab batch. The EMA requires the test batch to be at least 1/10th the size of a full production run or 100,000 units, whichever is larger. The FDA expects the same. Why? Because small-scale batches can behave differently. If the manufacturing process changes between the study batch and what’s sold in pharmacies, the results won’t mean anything. Companies often run pilot studies to fine-tune the formulation before the main trial. According to CRO insiders, using pilot data cuts failure rates in pivotal studies from 35% to under 10%.Step 3: Design the Study
Most bioequivalence studies use a crossover design. That means each volunteer takes both the generic and the brand-name drug, but in a random order. Half the group gets the generic first, then the brand. The other half gets the brand first, then the generic. This design controls for individual differences-like metabolism or body weight-because each person is their own control. The typical number of volunteers? Between 24 and 32 healthy adults. For drugs that vary a lot between people (like blood thinners or epilepsy meds), regulators allow more complex designs with four periods and 50 to 100 participants.Step 4: The Washout Period
After taking one drug, volunteers must wait before taking the other. This is called the washout period. It needs to be long enough for the first drug to leave the body completely. The rule? At least five half-lives of the drug. For a drug with a 12-hour half-life, that’s 60 hours. For one with a 72-hour half-life? That’s 360 hours-15 days. Underestimating this is one of the most common mistakes. One contract research organization lost $250,000 and three months because they used a 7-day washout for a drug with a 72-hour half-life. The leftover drug skewed the results, and the study had to be restarted.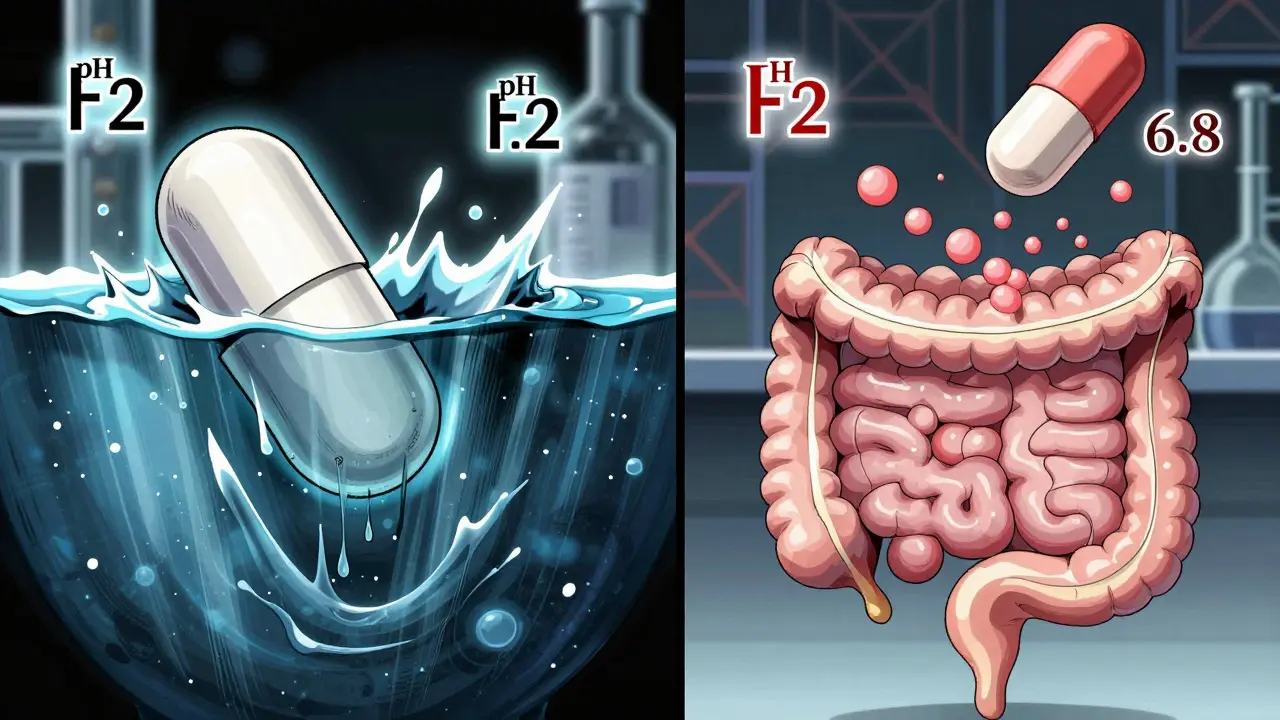
Step 5: Collect Blood Samples
Volunteers come in fasted. They swallow the pill with water, then sit quietly. Blood is drawn before the dose (baseline), then at specific times after. The standard schedule includes: pre-dose, one sample just before the expected peak concentration (Cmax), two around the peak, and three during the elimination phase. Sampling continues until the area under the curve (AUC) captures at least 80% of the total exposure. That usually means 3 to 5 half-lives of sampling. Blood is spun down to plasma, frozen, and shipped to a lab. The analytical method? Almost always liquid chromatography with tandem mass spectrometry (LC-MS/MS). It’s precise, sensitive, and can detect tiny amounts of the drug. The FDA requires precision within ±15%-±20% at the lowest detectable level.Step 6: Measure the Key Numbers
Two numbers matter most: Cmax and AUC. Cmax is the highest concentration of the drug in the blood. AUC is the total exposure over time-how much drug your body absorbed overall. These aren’t guesses. They’re calculated from the blood data using validated software. For extended-release pills, you might also look at AUC from 0 to 24 hours or other time points. For some drugs, like those that act locally in the gut, you might measure drug levels in stool instead of blood. But for most systemic drugs, plasma concentration is the gold standard.Step 7: The Statistical Test
The raw data isn’t enough. You need to compare the test and reference products. First, the Cmax and AUC values are log-transformed. Then, a statistical model (usually ANOVA) calculates the geometric mean ratio of test to reference. The result? A 90% confidence interval. For the study to pass, that interval must fall between 80% and 125% for both Cmax and AUC. That means the generic delivers 80% to 125% of the brand’s exposure. For narrow therapeutic index drugs-like warfarin or lithium-the window tightens to 90% to 111.11%. If it’s outside that range, the study fails. The FDA rejected Alembic’s generic Trulicity in 2022 because Cmax values varied too much across studies.Step 8: Dissolution Testing
Even if the blood results look good, regulators also check how the drug dissolves. You test both the generic and brand-name drug in different pH levels-starting with stomach acid (pH 1.2), then moving to intestinal fluid (pH 4.5, 6.8). You need at least 12 tablets or capsules per condition. The dissolution profiles must be similar. The f2 similarity factor must be above 50. If the generic dissolves too fast or too slow, it might not be absorbed the same way, even if blood levels look okay. This step is especially important for modified-release drugs.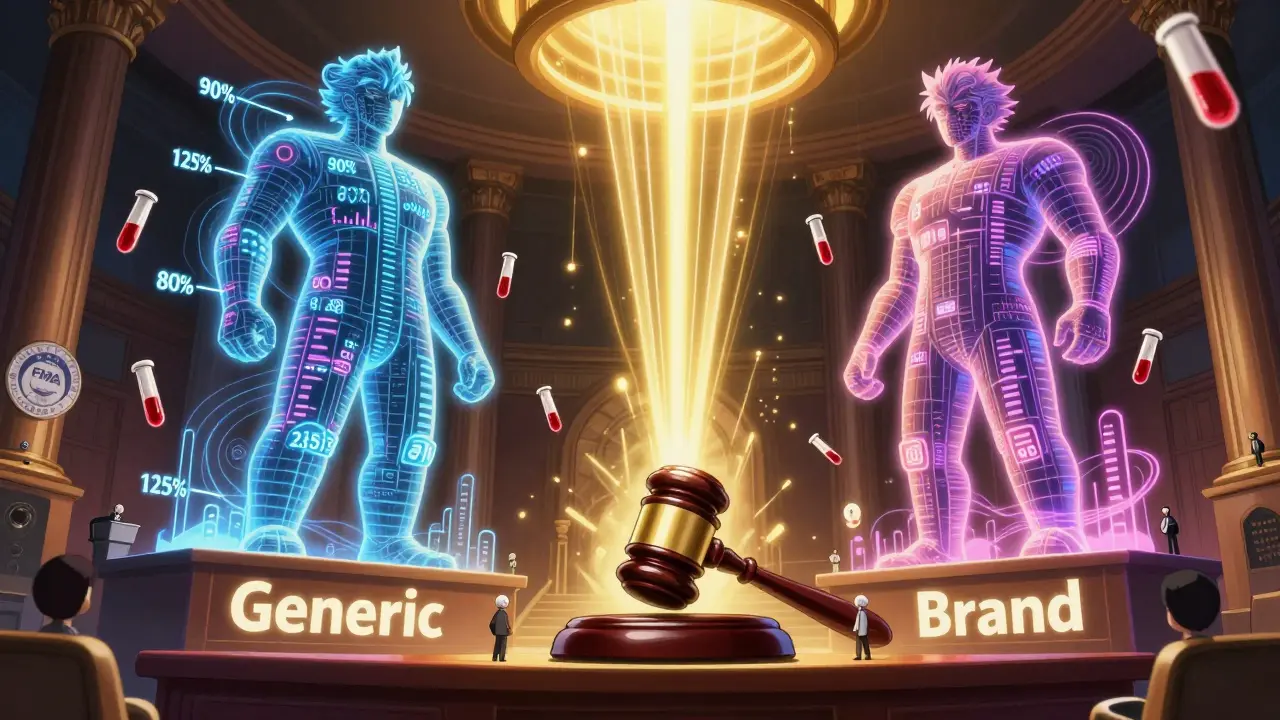
Step 9: Submit and Wait
Once the data is clean, you compile everything: the protocol, lab reports, statistical analysis, dissolution data, and subject records. You submit it to the FDA, EMA, or other agency. The FDA receives about 2,500 bioequivalence submissions a year. The average review time? Just over 10 months. Many applications get requests for more data. Common reasons: inadequate washout, missing time points, or statistical errors. The FDA says 45% of failed studies have washout issues, 30% have sampling problems, and 25% have statistical flaws.Alternatives and Exceptions
Not every drug needs a full bioequivalence study. For drugs with high solubility and permeability (BCS Class I), regulators may allow a waiver based on dissolution testing alone. That’s how 27% of 2022 approvals were granted without human trials. For complex products like inhalers, topical creams, or injectables, regulators require different approaches. The FDA now mandates clinical endpoint studies for some topical drugs. For drugs with extremely long half-lives (over two weeks), parallel studies are used-where one group gets the generic, another gets the brand. No crossover. No washout. Just direct comparison.What Happens When It Fails
Failure isn’t rare. About 1 in 5 bioequivalence studies don’t pass the first time. Common reasons: poor sampling timing, wrong statistical model, or analytical method issues. BioAgilytix reported that 22% of studies face delays due to problems with the lab assay. Each delay costs an average of $187,000. Companies that succeed invest in pilot studies, real-time data monitoring, and experienced teams. A good biostatistician knows how to handle replicate designs. A skilled bioanalyst ensures the LC-MS/MS method is robust. Clinical staff know how to manage washouts and subject compliance.The Bigger Picture
Bioequivalence studies are the backbone of generic drug access. They’re not perfect. Some experts warn that focusing only on blood levels might miss differences in how a drug acts in specific tissues-like the skin or lungs. That’s why the FDA now requires extra data for certain products. But for most systemic drugs, the system works. In 2022, the FDA approved 936 generic drugs based on bioequivalence data-98% of all generic approvals that year. The global market for these studies is growing fast, projected to hit $3.3 billion by 2030. As more blockbuster drugs lose patent protection, the demand for these studies will only rise. And the science behind them? It’s getting smarter. Modeling, simulation, and real-world data are slowly becoming part of the process. But for now, the blood test, the 90% confidence interval, and the 80-125% rule remain the standard.What happens if a bioequivalence study fails?
If a study fails, the company must revise the formulation, fix the protocol, or adjust the manufacturing process. They then run a new study. Common fixes include changing the drug’s excipients, adjusting the dissolution profile, or extending the washout period. Many companies run pilot studies before the main trial to catch issues early. Failure doesn’t mean the drug won’t ever be approved-it just means the first attempt didn’t meet the strict regulatory thresholds.
Are bioequivalence studies safe for participants?
Yes. Volunteers are healthy adults screened for medical conditions, medications, or habits that could interfere with the results. They’re monitored closely during the study, with medical staff on-site. Blood draws are minimal and done by trained professionals. Most studies last 10 to 20 days, and participants are compensated fairly. Dropout rates are typically between 5% and 15%, often due to personal reasons, not safety issues.
Why do some generics work differently for me than the brand?
Regulatory standards ensure generics are equivalent on average across a group of people. But individual biology varies. Factors like gut motility, liver enzymes, or even food intake can affect how a drug behaves in your body. If you notice a difference, talk to your doctor. It doesn’t mean the generic failed the study-it just means your body responds differently. Some people are more sensitive to small changes, especially with narrow therapeutic index drugs.
Can a generic drug be approved without human trials?
Yes, for certain drugs. If a drug is highly soluble and highly permeable (BCS Class I), and its dissolution profile matches the brand exactly, regulators may approve it based on in vitro testing alone. This is called a biowaiver. About 27% of generic approvals in 2022 used this route. It’s not allowed for complex formulations like extended-release pills, injectables, or topical products.
How long does a bioequivalence study take?
A typical crossover study takes 6 to 12 weeks from start to finish. That includes screening volunteers, dosing periods, washout, sampling, lab analysis, and data review. The actual time a volunteer spends in the clinic is usually 10 to 20 days. But the entire process-from protocol design to regulatory submission-can take 12 to 18 months, especially if pilot studies or revisions are needed.
Do all countries use the same bioequivalence standards?
Most major regulators-FDA, EMA, Health Canada, PMDA-follow similar principles, but there are differences. The FDA allows reference-scaled bioequivalence for highly variable drugs, while the EMA requires replicate crossover designs. Japan requires additional dissolution testing for some products. These variations mean a drug approved in the U.S. might need extra data to get approved in Europe or Japan. Harmonization efforts through ICH are reducing these gaps, but they still exist.
Chris & Kara Cutler
February 1, 2026 AT 01:14Rachel Liew
February 2, 2026 AT 22:59Jamie Allan Brown
February 4, 2026 AT 18:04Lisa Rodriguez
February 5, 2026 AT 01:22Ed Di Cristofaro
February 5, 2026 AT 06:26Lilliana Lowe
February 6, 2026 AT 01:44vivian papadatu
February 7, 2026 AT 23:52Deep Rank
February 9, 2026 AT 00:20Jaden Green
February 10, 2026 AT 03:31Angel Fitzpatrick
February 11, 2026 AT 07:55Nidhi Rajpara
February 11, 2026 AT 14:25